Chemists are often interested in developing new catalysts that will improve the efficiency of chemical reactions. They can look to nature to provide examples when designing these new materials. Metalloporphyrins are a class of metal complexes that appear a great deal in biological systems. These complexes, while they are effective catalysts, can decompose and oxidize unless they are protected somehow. A structure called a metal-organic framework (MOF) can provide this protection. The MOF includes catalytically active sites within the structure of a porous material composed of organic linkers that bridge modular metal clusters that act as structural nodes. MOFs are useful for a wide variety of chemical applications.
In a recent paper, members of Professor Chris Cramer’s MSI research group, in collaboration with researchers at Northwestern University, Argonne National Laboratory, Purdue University, and King Abdulaziz University, reported that they had developed a novel MOF with porphyrinic struts and hafnium-based nodes. The research involved experimental work as well as density functional theory (DFT) calculations. (DFT is a computational quantum mechanical modeling method that can be used to investigate the electronic structures of materials.) In this work, theory played a critical role in identifying the creation of unexpected catalytic sites that led to reactivity both new and complementary to existing catalysts for the generation of chemically important β-aminoalcohols. The paper was published in the Journal of the American Chemical Society (Cramer group members are in bold): Beyzavi, M. Hassan, Nicolaas A. Vermeulen, Ashlee J. Howarth, Samat Tussupbayev, Aaron B. League, Neil M. Schweitzer, James R. Gallagher, et al. [Christopher Cramer] 2015. A hafnium-based metal-organic framework as a nature-inspired tandem reaction catalyst. Journal of the American Chemical Society 137 (42) (OCT 28): 13624-31. Dr. Samat Tussupbayev is a post-doc and Aaron League is a graduate student.
Professor Cramer is a long-time Principal Investigator at MSI. His research involves a wide-ranging number of projects that use novel and/or established classical and quantum mechanical methodologies to model chemical structures, properties, and reactivities. More information about his group’s work can be found on the Cramer Group website. Professor Cramer is also a member of the Chemical Theory Center, whose other members include several MSI PIs: Regents Professor Donald Truhlar, Professor Laura Gagliardi, Professor J. Ilja Siepmann, and Professor Jiali Gao. Previous work by the Cramer group was featured in a Research Spotlight in May 2014.
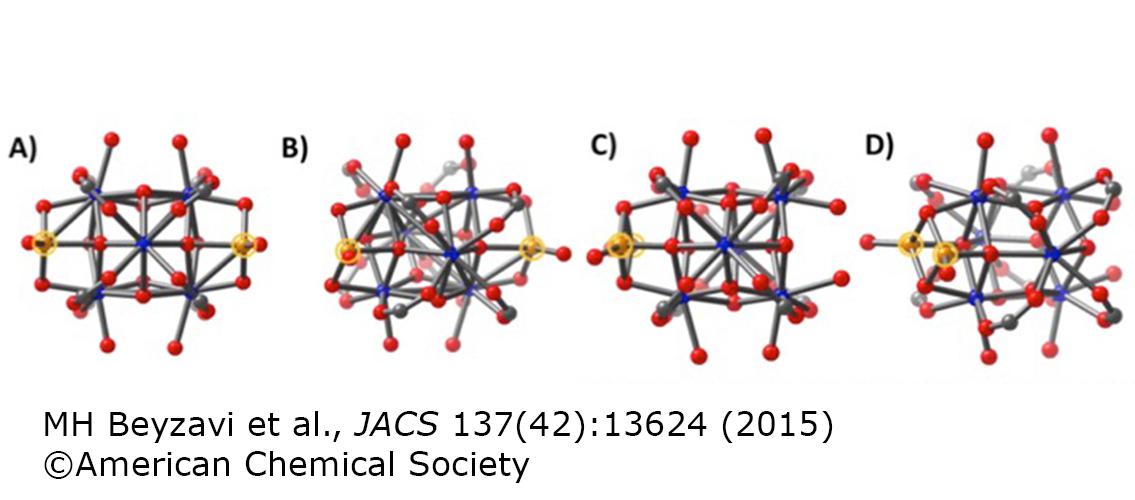
Image description: Ball-and-stick representations of truncated clusters for the lowest-energy computationally determined Hf node containing one Fe atom on each of two distinct faces (A and B, which are perspectives differing by a 30 deg rotation about the vertical axis) and the same Hf node having instead two bridged Fe atoms sited on a single face (C and D, again showing different perspectives). Carbon, oxygen, hafnium, and iron are represented by gray, red, blue, and orange spheres, respectively. The Fe atoms are highlighted with orange circles for emphasis; to improve clarity, H atoms are not shown. Image and description from M. Hassan Beyzavi et al., J. Am. Chem. Soc. 2015, 137, 13624-13631, DOI: 10.1021/jacs.5b08440. © 2015 American Chemical Society
posted on December 16, 2015